






The growing proportion of the elderly population represents an increasing socioeconomic challenge, not least because of age-associated diseases. It is therefore increasingly pertinent to find interventions for age-associated diseases such as Alzheimer's, Parkinson's and cardiovascular diseases. Although the cause of aging is currently unknown accumulation of damage to our genome, the DNA, may be an contributing factor.
In the Scheibye-Knudsen lab we try to understand the cellular and organismal consequences of DNA damage with the aim of developing interventions. We have discovered that DNA damage leads to changes in certain metabolites and that replenishment of these molecules may alter the rate of aging in model organisms. These findings suggest that normal aging and age-associated diseases may be malleable to similar interventions. The hope is to develop interventions that will allow everyone to live healthier, happier and more productive lives.
Monogenic Diseases of DNA Repair.
Guido Keijzers, Daniela Bakula, Morten Scheibye-Knudsen,
The New England journal of medicine - 11 2017
[abstract]
Mitophagy in neurodegeneration and aging.
Elayne M Fivenson, Sofie Lautrup, Nuo Sun, Morten Scheibye-Knudsen, Tinna Stevnsner, Hilde Nilsen, Vilhelm A Bohr, Evandro F Fang,
Neurochemistry international - Oct 2017
[abstract]
Mitochondrial dysfunction contributes to normal aging and a wide spectrum of age-related diseases, including neurodegenerative disorders such as Parkinson's disease and Alzheimer's disease. It is important to maintain a healthy mitochondrial population which is tightly regulated by proteolysis and mitophagy. Mitophagy is a specialized form of autophagy that regulates the turnover of damaged and dysfunctional mitochondria, organelles that function in producing energy for the cell in the form of ATP and regulating energy homeostasis. Mechanistic studies on mitophagy across species highlight a sophisticated and integrated cellular network that regulates the degradation of mitochondria. Strategies directed at maintaining a healthy mitophagy level in aged individuals might have beneficial effects. In this review, we provide an updated mechanistic overview of mitophagy pathways and discuss the role of reduced mitophagy in neurodegeneration. We also highlight potential translational applications of mitophagy-inducing compounds, such as NAD+ precursors and urolithins.
Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway.
Evandro F Fang, Tyler B Waltz, Henok Kassahun, Qiping Lu, Jesse S Kerr, Marya Morevati, Elayne M Fivenson, Bradley N Wollman, Krisztina Marosi, Mark A Wilson, Wendy B Iser, D Mark Eckley, Yongqing Zhang, Elin Lehrmann, Ilya G Goldberg, Morten Scheibye-Knudsen, Mark P Mattson, Hilde Nilsen, Vilhelm A Bohr, Kevin G Becker,
Scientific reports - Apr 2017
[abstract]
Aging is a major international concern that brings formidable socioeconomic and healthcare challenges. Small molecules capable of improving the health of older individuals are being explored. Small molecules that enhance cellular stress resistance are a promising avenue to alleviate declines seen in human aging. Tomatidine, a natural compound abundant in unripe tomatoes, inhibits age-related skeletal muscle atrophy in mice. Here we show that tomatidine extends lifespan and healthspan in C. elegans, an animal model of aging which shares many major longevity pathways with mammals. Tomatidine improves many C. elegans behaviors related to healthspan and muscle health, including increased pharyngeal pumping, swimming movement, and reduced percentage of severely damaged muscle cells. Microarray, imaging, and behavioral analyses reveal that tomatidine maintains mitochondrial homeostasis by modulating mitochondrial biogenesis and PINK-1/DCT-1-dependent mitophagy. Mechanistically, tomatidine induces mitochondrial hormesis by mildly inducing ROS production, which in turn activates the SKN-1/Nrf2 pathway and possibly other cellular antioxidant response pathways, followed by increased mitophagy. This mechanism occurs in C. elegans, primary rat neurons, and human cells. Our data suggest that tomatidine may delay some physiological aspects of aging, and points to new approaches for pharmacological interventions for diseases of aging.
In vivo Mitophagy Monitoring in Caenorhabditis elegans to Determine Mitochondrial Homeostasis.
Konstantinos Palikaras, Nektarios Tavernarakis,
Bio-protocol - Apr 2017
[abstract]
Perturbation of mitochondrial function is a major hallmark of several pathological conditions and ageing, underlining the essential role of fine-tuned mitochondrial activity (Lopez-Otin et al., 2013). Mitochondrial selective autophagy, known as mitophagy, mediates the removal of dysfunctional and/or superfluous organelles, preserving cellular and organismal homeostasis (Palikaras and Tavernarakis, 2014; Pickrell and Youle, 2015; Scheibye-Knudsen et al., 2015). In this protocol, we describe a method for assessing mitophagy in the nematode Caenorhabditis elegans.
Cockayne syndrome: Clinical features, model systems and pathways.
Ajoy C Karikkineth, Morten Scheibye-Knudsen, Elayne Fivenson, Deborah L Croteau, Vilhelm A Bohr,
Ageing research reviews - Jan 2017
[abstract]
Cockayne syndrome (CS) is a disorder characterized by a variety of clinical features including cachectic dwarfism, severe neurological manifestations including microcephaly and cognitive deficits, pigmentary retinopathy, cataracts, sensorineural deafness, and ambulatory and feeding difficulties, leading to death by 12 years of age on average. It is an autosomal recessive disorder, with a prevalence of approximately 2.5 per million. There are several phenotypes (1-3) and two complementation groups (CSA and CSB), and CS overlaps with xeroderma pigmentosum (XP). It has been considered a progeria, and many of the clinical features resemble accelerated aging. As such, the study of CS affords an opportunity to better understand the underlying mechanisms of aging. The molecular basis of CS has traditionally been ascribed to defects in transcription and transcription-coupled nucleotide excision repair (TC-NER). However, recent work suggests that defects in base excision DNA repair and mitochondrial functions may also play key roles. This opens up the possibility for molecular interventions in CS, and by extrapolation, possibly in aging.
3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons.
Krisztina Marosi, Sang Woo Kim, Keelin Moehl, Morten Scheibye-Knudsen, Aiwu Cheng, Roy Cutler, Simonetta Camandola, Mark P Mattson,
Journal of neurochemistry - 12 2016
[abstract]
During fasting and vigorous exercise, a shift of brain cell energy substrate utilization from glucose to the ketone 3-hydroxybutyrate (3OHB) occurs. Studies have shown that 3OHB can protect neurons against excitotoxicity and oxidative stress, but the underlying mechanisms remain unclear. Neurons maintained in the presence of 3OHB exhibited increased oxygen consumption and ATP production, and an elevated NAD+ /NADH ratio. We found that 3OHB metabolism increases mitochondrial respiration which drives changes in expression of brain-derived neurotrophic factor (BDNF) in cultured cerebral cortical neurons. The mechanism by which 3OHB induces Bdnf gene expression involves generation of reactive oxygen species, activation of the transcription factor NF-κB, and activity of the histone acetyltransferase p300/EP300. Because BDNF plays important roles in synaptic plasticity and neuronal stress resistance, our findings suggest cellular signaling mechanisms by which 3OHB may mediate adaptive responses of neurons to fasting, exercise, and ketogenic diets.
A ketogenic diet accelerates neurodegeneration in mice with induced mitochondrial DNA toxicity in the forebrain.
Knut H Lauritzen, Md Mahdi Hasan-Olive, Christine E Regnell, Liv Kleppa, Morten Scheibye-Knudsen, Albert Gjedde, Arne Klungland, Vilhelm A Bohr, Jon Storm-Mathisen, Linda H Bergersen,
Neurobiology of aging - Dec 2016
[abstract]
Mitochondrial genome maintenance plays a central role in preserving brain health. We previously demonstrated accumulation of mitochondrial DNA damage and severe neurodegeneration in transgenic mice inducibly expressing a mutated mitochondrial DNA repair enzyme (mutUNG1) selectively in forebrain neurons. Here, we examine whether severe neurodegeneration in mutUNG1-expressing mice could be rescued by feeding the mice a ketogenic diet, which is known to have beneficial effects in several neurological disorders. The diet increased the levels of superoxide dismutase 2, and mitochondrial mass, enzymes, and regulators such as SIRT1 and FIS1, and appeared to downregulate N-methyl-D-aspartic acid (NMDA) receptor subunits NR2A/B and upregulate γ-aminobutyric acid A (GABAA) receptor subunits α1. However, unexpectedly, the ketogenic diet aggravated neurodegeneration and mitochondrial deterioration. Electron microscopy showed structurally impaired mitochondria accumulating in neuronal perikarya. We propose that aggravation is caused by increased mitochondrial biogenesis of generally dysfunctional mitochondria. This study thereby questions the dogma that a ketogenic diet is unambiguously beneficial in mitochondrial disorders.
Neurodegeneration in accelerated aging.
Moren Scheibye-Knudsen,
Danish medical journal - Nov 2016
[abstract]
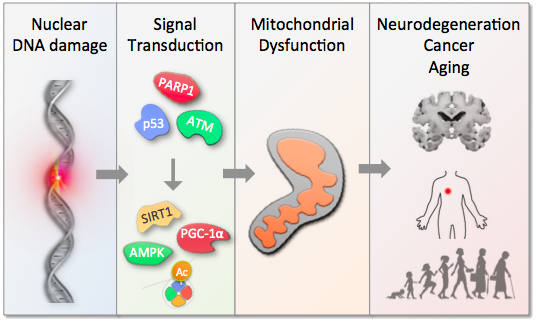
The growing proportion of elderly people represents an increasing economic burden, not least because of age-associated diseases that pose a significant cost to the health service. Finding possible interventions to age-associated disorders therefore have wide ranging implications. A number of genetically defined accelerated aging diseases have been characterized that can aid in our understanding of aging. Interestingly, all these diseases are associated with defects in the maintenance of our genome. A subset of these disorders, Cockayne syndrome, Xeroderma pigmentosum group A and ataxia-telangiectasia, show neurological involvement reminiscent of what is seen in primary human mitochondrial diseases. Mitochondria are the power plants of the cells converting energy stored in oxygen, sugar, fat, and protein into ATP, the energetic currency of our body. Emerging evidence has linked this organelle to aging and finding mitochondrial dysfunction in accelerated aging disorders thereby strengthens the mitochondrial theory of aging. This theory states that an accumulation of damage to the mitochondria may underlie the process of aging. Indeed, it appears that some accelerated aging disorders that show neurodegeneration also have mitochondrial dysfunction. The mitochondrial alterations may be secondary to defects in nuclear DNA repair. Indeed, nuclear DNA damage may lead to increased energy consumption, alterations in mitochondrial ATP production and defects in mitochondrial recycling, a term called mitophagy. These changes may be caused by activation of poly-ADP-ribose-polymerase 1 (PARP1), an enzyme that responds to DNA damage. Upon activation PARP1 utilizes key metabolites that attenuate pathways that are normally protective for the cell. Notably, pharmacological inhibition of PARP1 or reconstitution of the metabolites rescues the changes caused by PARP1 hyperactivation and in many cases reverse the phenotypes associated with accelerated aging. This implies that modulation of PARP1 or the downstream metabolites may be a therapeutic strategy for treating accelerated aging disorders and potentially age-associated neurological decline seen in the normal population.
Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA.
Morten Scheibye-Knudsen, Anne Tseng, Martin Borch Jensen, Karsten Scheibye-Alsing, Evandro Fei Fang, Teruaki Iyama, Sanjay Kumar Bharti, Krisztina Marosi, Lynn Froetscher, Henok Kassahun, David Mark Eckley, Robert W Maul, Paul Bastian, Supriyo De, Soumita Ghosh, Hilde Nilsen, Ilya G Goldberg, Mark P Mattson, David M Wilson, Robert M Brosh, Myriam Gorospe, Vilhelm A Bohr,
Proceedings of the National Academy of Sciences of the United States of America - Nov 2016
[abstract]
Cockayne syndrome is a neurodegenerative accelerated aging disorder caused by mutations in the CSA or CSB genes. Although the pathogenesis of Cockayne syndrome has remained elusive, recent work implicates mitochondrial dysfunction in the disease progression. Here, we present evidence that loss of CSA or CSB in a neuroblastoma cell line converges on mitochondrial dysfunction caused by defects in ribosomal DNA transcription and activation of the DNA damage sensor poly-ADP ribose polymerase 1 (PARP1). Indeed, inhibition of ribosomal DNA transcription leads to mitochondrial dysfunction in a number of cell lines. Furthermore, machine-learning algorithms predict that diseases with defects in ribosomal DNA (rDNA) transcription have mitochondrial dysfunction, and, accordingly, this is found when factors involved in rDNA transcription are knocked down. Mechanistically, loss of CSA or CSB leads to polymerase stalling at non-B DNA in a neuroblastoma cell line, in particular at G-quadruplex structures, and recombinant CSB can melt G-quadruplex structures. Indeed, stabilization of G-quadruplex structures activates PARP1 and leads to accelerated aging in Caenorhabditis elegans In conclusion, this work supports a role for impaired ribosomal DNA transcription in Cockayne syndrome and suggests that transcription-coupled resolution of secondary structures may be a mechanism to repress spurious activation of a DNA damage response.
See more
NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair.
Evandro Fei Fang, Henok Kassahun, Deborah L Croteau, Morten Scheibye-Knudsen, Krisztina Marosi, Huiming Lu, Raghavendra A Shamanna, Sumana Kalyanasundaram, Ravi Chand Bollineni, Mark A Wilson, Wendy B Iser, Bradley N Wollman, Marya Morevati, Jun Li, Jesse S Kerr, Qiping Lu, Tyler B Waltz, Jane Tian, David A Sinclair, Mark P Mattson, Hilde Nilsen, Vilhelm A Bohr,
Cell metabolism - Oct 2016
[abstract]
Ataxia telangiectasia (A-T) is a rare autosomal recessive disease characterized by progressive neurodegeneration and cerebellar ataxia. A-T is causally linked to defects in ATM, a master regulator of the response to and repair of DNA double-strand breaks. The molecular basis of cerebellar atrophy and neurodegeneration in A-T patients is unclear. Here we report and examine the significance of increased PARylation, low NAD+, and mitochondrial dysfunction in ATM-deficient neurons, mice, and worms. Treatments that replenish intracellular NAD+ reduce the severity of A-T neuropathology, normalize neuromuscular function, delay memory loss, and extend lifespan in both animal models. Mechanistically, treatments that increase intracellular NAD+ also stimulate neuronal DNA repair and improve mitochondrial quality via mitophagy. This work links two major theories on aging, DNA damage accumulation, and mitochondrial dysfunction through nuclear DNA damage-induced nuclear-mitochondrial signaling, and demonstrates that they are important pathophysiological determinants in premature aging of A-T, pointing to therapeutic interventions.
Cytochrome b5 reductase and the control of lipid metabolism and healthspan.
Alejandro Martin-Montalvo, Yaning Sun, Alberto Diaz-Ruiz, Ahmed Ali, Vincent Gutierrez, Hector H Palacios, Jessica Curtis, Emilio Siendones, Julia Ariza, Gelareh A Abulwerdi, Xiaoping Sun, Annie X Wang, Kevin J Pearson, Kenneth W Fishbein, Richard G Spencer, Miao Wang, Xianlin Han, Morten Scheibye-Knudsen, Joe A Baur, Howard G Shertzer, Placido Navas, Jose Manuel Villalba, Sige Zou, Michel Bernier, Rafael de Cabo,
NPJ aging and mechanisms of disease - 2016
[abstract]
Cytochrome b5 reductases (CYB5R) are required for the elongation and desaturation of fatty acids, cholesterol synthesis and mono-oxygenation of cytochrome P450 enzymes, all of which are associated with protection against metabolic disorders. However, the physiological role of CYB5R in the context of metabolism, healthspan and aging remains ill-defined. We generated CYB5R-overexpressing flies (CYB5R-OE) and created a transgenic mouse line overexpressing CYB5R3 (CYB5R3-Tg) in the C57BL/6J background to investigate the function of this class of enzymes as regulators of metabolism and age-associated pathologies. Gender- and/or stage-specific induction of CYB5R, and pharmacological activation of CYB5R with tetrahydroindenoindole extended fly lifespan. Increased expression of CYB5R3 was associated with significant improvements in several metabolic parameters that resulted in modest lifespan extension in mice. Diethylnitrosamine-induced liver carcinogenesis was reduced in CYB5R3-Tg mice. Accumulation of high levels of long-chain polyunsaturated fatty acids, improvement in mitochondrial function, decrease in oxidative damage and inhibition of chronic pro-inflammatory pathways occurred in the transgenic animals. These results indicate that CYB5R represents a new target in the study of genes that regulate lipid metabolism and healthspan.
Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice.
Sarah J Mitchell, Julio Madrigal-Matute, Morten Scheibye-Knudsen, Evandro Fang, Miguel Aon, José A González-Reyes, Sonia Cortassa, Susmita Kaushik, Marta Gonzalez-Freire, Bindi Patel, Devin Wahl, Ahmed Ali, Miguel Calvo-Rubio, María I Burón, Vincent Guiterrez, Theresa M Ward, Hector H Palacios, Huan Cai, David W Frederick, Christopher Hine, Filomena Broeskamp, Lukas Habering, John Dawson, T Mark Beasley, Junxiang Wan, Yuji Ikeno, Gene Hubbard, Kevin G Becker, Yongqing Zhang, Vilhelm A Bohr, Dan L Longo, Placido Navas, Luigi Ferrucci, David A Sinclair, Pinchas Cohen, Josephine M Egan, James R Mitchell, Joseph A Baur, David B Allison, R Michael Anson, José M Villalba, Frank Madeo, Ana Maria Cuervo, Kevin J Pearson, Donald K Ingram, Michel Bernier, Rafael de Cabo,
Cell metabolism - Jun 2016
[abstract]
Calorie restriction (CR) is the most robust non-genetic intervention to delay aging. However, there are a number of emerging experimental variables that alter CR responses. We investigated the role of sex, strain, and level of CR on health and survival in mice. CR did not always correlate with lifespan extension, although it consistently improved health across strains and sexes. Transcriptional and metabolomics changes driven by CR in liver indicated anaplerotic filling of the Krebs cycle together with fatty acid fueling of mitochondria. CR prevented age-associated decline in the liver proteostasis network while increasing mitochondrial number, preserving mitochondrial ultrastructure and function with age. Abrogation of mitochondrial function negated life-prolonging effects of CR in yeast and worms. Our data illustrate the complexity of CR in the context of aging, with a clear separation of outcomes related to health and survival, highlighting complexities of translation of CR into human interventions.
Nuclear DNA damage signalling to mitochondria in ageing.
Evandro Fei Fang, Morten Scheibye-Knudsen, Katrin F Chua, Mark P Mattson, Deborah L Croteau, Vilhelm A Bohr,
Nature reviews. Molecular cell biology - 05 2016
[abstract]
Mitochondrial dysfunction is a hallmark of ageing, and mitochondrial maintenance may lead to increased healthspan. Emerging evidence suggests a crucial role for signalling from the nucleus to mitochondria (NM signalling) in regulating mitochondrial function and ageing. An important initiator of NM signalling is nuclear DNA damage, which accumulates with age and may contribute to the development of age-associated diseases. DNA damage-dependent NM signalling constitutes a network that includes nuclear sirtuins and controls genomic stability and mitochondrial integrity. Pharmacological modulation of NM signalling is a promising novel approach for the prevention and treatment of age-associated diseases.
A research agenda for aging in China in the 21st century.
Evandro Fei Fang, Morten Scheibye-Knudsen, Heiko J Jahn, Juan Li, Li Ling, Hongwei Guo, Xinqiang Zhu, Victor Preedy, Huiming Lu, Vilhelm A Bohr, Wai Yee Chan, Yuanli Liu, Tzi Bun Ng,
Ageing research reviews - Nov 2015
[abstract]
China is encountering formidable healthcare challenges brought about by the problem of aging. By 2050, there will be 400 million Chinese citizens aged 65+, 150 million of whom will be 80+. The undesirable consequences of the one-child policy, rural-to-urban migration, and expansion of the population of 'empty nest' elders are eroding the traditional family care of the elders, further exacerbating the burden borne by the current public healthcare system. The challenges of geriatric care demand prompt attention by proposing strategies for improvement in several key areas. Major diseases of the elderly that need more attention include chronic non-communicable diseases and mental health disorders. We suggest the establishment of a home care-dominated geriatric care system, and a proactive role for researchers on aging in reforming geriatric care through policy dialogs. We propose ideas for preparation of the impending aging burden and the creation of a nurturing environment conducive to healthy aging in China.
DNA Damage, DNA Repair, Aging, and Neurodegeneration.
Scott Maynard, Evandro Fei Fang, Morten Scheibye-Knudsen, Deborah L Croteau, Vilhelm A Bohr,
Cold Spring Harbor perspectives in medicine - Sep 2015
[abstract]
Aging in mammals is accompanied by a progressive atrophy of tissues and organs, and stochastic damage accumulation to the macromolecules DNA, RNA, proteins, and lipids. The sequence of the human genome represents our genetic blueprint, and accumulating evidence suggests that loss of genomic maintenance may causally contribute to aging. Distinct evidence for a role of imperfect DNA repair in aging is that several premature aging syndromes have underlying genetic DNA repair defects. Accumulation of DNA damage may be particularly prevalent in the central nervous system owing to the low DNA repair capacity in postmitotic brain tissue. It is generally believed that the cumulative effects of the deleterious changes that occur in aging, mostly after the reproductive phase, contribute to species-specific rates of aging. In addition to nuclear DNA damage contributions to aging, there is also abundant evidence for a causative link between mitochondrial DNA damage and the major phenotypes associated with aging. Understanding the mechanistic basis for the association of DNA damage and DNA repair with aging and age-related diseases, such as neurodegeneration, would give insight into contravening age-related diseases and promoting a healthy life span.
A novel method for determining human ex vivo submaximal skeletal muscle mitochondrial function.
Martin Hey-Mogensen, Martin Gram, Martin Borch Jensen, Michael Taulo Lund, Christina Neigaard Hansen, Morten Scheibye-Knudsen, Vilhelm A Bohr, Flemming Dela,
The Journal of physiology - Sep 2015
[abstract]
The present study utilized a novel method aiming to investigate mitochondrial function in human skeletal muscle at submaximal levels and at a predefined membrane potential. The effect of age and training status was investigated using a cross-sectional design. Ageing was found to be related to decreased leak regardless of training status. Increased training status was associated with increased mitochondrial hydrogen peroxide emission. Despite numerous studies, there is no consensus about whether mitochondrial function is altered with increased age. The novelty of the present study is the determination of mitochondrial function at submaximal activity rates, which is more physiologically relevant than the ex vivo functionality protocols used previously. Muscle biopsies were taken from 64 old or young male subjects (aged 60-70 or 20-30 years). Aged subjects were recruited as trained or untrained. Muscle biopsies were used for the isolation of mitochondria and subsequent measurements of DNA repair, anti-oxidant capacity and mitochondrial protein levels (complexes I-V). Mitochondrial function was determined by simultaneous measurement of oxygen consumption, membrane potential and hydrogen peroxide emission using pyruvate + malate (PM) or succinate + rotenone (SR) as substrates. Proton leak was lower in aged subjects when determined at the same membrane potential and was unaffected by training status. State 3 respiration was lower in aged untrained subjects. This effect, however, was alleviated in aged trained subjects. H2 O2 emission with PM was higher in aged subjects, and was exacerbated by training, although it was not changed when using SR. However, with a higher manganese superoxide dismuthase content, the trained aged subjects may actually have lower or similar mitochondrial superoxide emission compared to the untrained subjects. We conclude that ageing and the physical activity level in aged subjects are both related to changes in the intrinsic functionality of the mitochondrion in skeletal muscle. Both of these changes could be important factors in determining the metabolic health of the aged skeletal muscle cell.
Animal models of aging research: implications for human aging and age-related diseases.
Sarah J Mitchell, Morten Scheibye-Knudsen, Dan L Longo, Rafael de Cabo,
Annual review of animal biosciences - 2015
[abstract]
Aging is characterized by an increasing morbidity and functional decline that eventually results in the death of an organism. Aging is the largest risk factor for numerous human diseases, and understanding the aging process may thereby facilitate the development of new treatments for age-associated diseases. The use of humans in aging research is complicated by many factors, including ethical issues; environmental and social factors; and perhaps most importantly, their long natural life span. Although cellular models of human disease provide valuable mechanistic information, they are limited in that they may not replicate the in vivo biology. Almost all organisms age, and thus animal models can be useful for studying aging. Herein, we review some of the major models currently used in aging research and discuss their benefits and pitfalls, including interventions known to extend life span and health span. Finally, we conclude by discussing the future of animal models in aging research.
Protecting the mitochondrial powerhouse.
Morten Scheibye-Knudsen, Evandro F Fang, Deborah L Croteau, David M Wilson, Vilhelm A Bohr,
Trends in cell biology - Mar 2015
[abstract]
Mitochondria are the oxygen-consuming power plants of cells. They provide a critical milieu for the synthesis of many essential molecules and allow for highly efficient energy production through oxidative phosphorylation. The use of oxygen is, however, a double-edged sword that on the one hand supplies ATP for cellular survival, and on the other leads to the formation of damaging reactive oxygen species (ROS). Different quality control pathways maintain mitochondria function including mitochondrial DNA (mtDNA) replication and repair, fusion-fission dynamics, free radical scavenging, and mitophagy. Further, failure of these pathways may lead to human disease. We review these pathways and propose a strategy towards a treatment for these often untreatable disorders.
Loss of NEIL1 causes defects in olfactory function in mice.
Chandrika Canugovi, Magdalena Misiak, Morten Scheibye-Knudsen, Deborah L Croteau, Mark P Mattson, Vilhelm A Bohr,
Neurobiology of aging - Feb 2015
[abstract]
Oxidative DNA damage accumulation has been implicated in neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease. The base excision repair pathway is a primary responder to oxidative DNA damage. Effects of loss of base excision repair on normal brain function is a relatively nascent area of research that needs further exploration for better understanding of related brain diseases. Recently, we found that loss of a versatile DNA glycosylase endonuclease 8-like 1 (NEIL1) causes deficits in spatial memory retention using the Morris water maze test. Furthermore, we found that there is a significant loss of NEIL1 enzyme levels and its activity in postmortem Alzheimer's disease brains. Based on the Allen Brain Atlas in situ hybridization data, the expression levels of Neil1 messenger RNA are higher in the olfactory bulb compared with other areas of the brain. Olfaction in mice is a central brain function that involves many central nervous system pathways. Here, we studied the effect of complete loss of Neil1 gene on olfactory function. We explored olfactory function in mice with 3 different behavioral tests namely, olfactory sensitivity, performance, and buried food tests. Neil1(-/-) mice performed poorly compared with wild-type mice in all 3 tests. Our data indicate that loss of Neil1 causes olfactory function deficits supporting our previous findings and that normal brain function requires robust DNA repair.
A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome.
Morten Scheibye-Knudsen, Sarah J Mitchell, Evandro F Fang, Teruaki Iyama, Theresa Ward, James Wang, Christopher A Dunn, Nagendra Singh, Sebastian Veith, Md Mahdi Hasan-Olive, Aswin Mangerich, Mark A Wilson, Mark P Mattson, Linda H Bergersen, Victoria C Cogger, Alessandra Warren, David G Le Couteur, Ruin Moaddel, David M Wilson, Deborah L Croteau, Rafael de Cabo, Vilhelm A Bohr,
Cell metabolism - Nov 2014
[abstract]
Cockayne syndrome (CS) is an accelerated aging disorder characterized by progressive neurodegeneration caused by mutations in genes encoding the DNA repair proteins CS group A or B (CSA or CSB). Since dietary interventions can alter neurodegenerative processes, Csb(m/m) mice were given a high-fat, caloric-restricted, or resveratrol-supplemented diet. High-fat feeding rescued the metabolic, transcriptomic, and behavioral phenotypes of Csb(m/m) mice. Furthermore, premature aging in CS mice, nematodes, and human cells results from aberrant PARP activation due to deficient DNA repair leading to decreased SIRT1 activity and mitochondrial dysfunction. Notably, β-hydroxybutyrate levels are increased by the high-fat diet, and β-hydroxybutyrate, PARP inhibition, or NAD(+) supplementation can activate SIRT1 and rescue CS-associated phenotypes. Mechanistically, CSB can displace activated PARP1 from damaged DNA to limit its activity. This study connects two emerging longevity metabolites, β-hydroxybutyrate and NAD(+), through the deacetylase SIRT1 and suggests possible interventions for CS.
Di-(2-ethylhexyl) phthalate inhibits DNA replication leading to hyperPARylation, SIRT1 attenuation, and mitochondrial dysfunction in the testis.
Xiaolin Li, Evandro Fei Fang, Morten Scheibye-Knudsen, Honghua Cui, Lu Qiu, Jian Li, Yuping He, Jing Huang, Vilhelm A Bohr, Tzi Bun Ng, Hongwei Guo,
Scientific reports - Sep 2014
[abstract]
Di-(2-ethylhexyl)-phthalate (DEHP) is a ubiquitously used endocrine disruptor.There is widespread exposure to DEHP in the general population which has raised substantial public concern due to its potential detrimental health effects. It is particularly pertinent to investigate the molecular mechanisms of its testicular toxicity which are largely unknown. By feeding male rats DEHP for 2 weeks, rat spermatogenesis became disrupted, resulting in a decreased number of spermatocytes and spermatids. Since rapidly dividing tissues appeared to be particularly vulnerable to DEHP toxicity we investigated the effect of DEHP on DNA replication. Intriguingly, DEHP appeared to inhibit DNA replication as evidenced by results of fiber tract analysis. This led to induction of the mitochondrial apoptotic pathways and increased ROS production. Furthermore, the toxicity of DEHP led to respiratory chain defects and attenuation of ATP level probably brought about by hyperPARylation and undermined SIRT1 activity. Our findings reveal a previously unknown mitochondrial dysfunction in DEHP-induced testicular toxicity and highlight the importance of SIRT1 in male reproduction.
Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders.
Morten Scheibye-Knudsen, Evandro Fei Fang, Deborah L Croteau, Vilhelm A Bohr,
Autophagy - Aug 2014
[abstract]
DNA repair is a prerequisite for life as we know it, and defects in DNA repair lead to accelerated aging. Xeroderma pigmentosum group A (XPA) is a classic DNA repair-deficient disorder with patients displaying sun sensitivity and cancer susceptibility. XPA patients also exhibit neurodegeneration, leading to cerebellar atrophy, neuropathy, and hearing loss, through a mechanism that has remained elusive. Using in silico, in vitro, and in vivo studies, we discovered defective mitophagy in XPA due to PARP1 hyperactivation and NAD(+) (and thus, SIRT1) depletion. This leads to mitochondrial membrane hyper-polarization, PINK1 cleavage and defective mitophagy. This study underscores the importance of mitophagy in promoting a healthy pool of mitochondria and in preventing neurodegeneration and premature aging.
SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass.
Evi M Mercken, Sarah J Mitchell, Alejandro Martin-Montalvo, Robin K Minor, Maria Almeida, Ana P Gomes, Morten Scheibye-Knudsen, Hector H Palacios, Jordan J Licata, Yongqing Zhang, Kevin G Becker, Husam Khraiwesh, José A González-Reyes, José M Villalba, Joseph A Baur, Peter Elliott, Christoph Westphal, George P Vlasuk, James L Ellis, David A Sinclair, Michel Bernier, Rafael de Cabo,
Aging cell - Oct 2014
[abstract]
Increased expression of SIRT1 extends the lifespan of lower organisms and delays the onset of age-related diseases in mammals. Here, we show that SRT2104, a synthetic small molecule activator of SIRT1, extends both mean and maximal lifespan of mice fed a standard diet. This is accompanied by improvements in health, including enhanced motor coordination, performance, bone mineral density, and insulin sensitivity associated with higher mitochondrial content and decreased inflammation. Short-term SRT2104 treatment preserves bone and muscle mass in an experimental model of atrophy. These results demonstrate it is possible to design a small molecule that can slow aging and delay multiple age-related diseases in mammals, supporting the therapeutic potential of SIRT1 activators in humans.
Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction.
Evandro Fei Fang, Morten Scheibye-Knudsen, Lear E Brace, Henok Kassahun, Tanima SenGupta, Hilde Nilsen, James R Mitchell, Deborah L Croteau, Vilhelm A Bohr,
Cell - May 2014
[abstract]
Mitochondrial dysfunction is a common feature in neurodegeneration and aging. We identify mitochondrial dysfunction in xeroderma pigmentosum group A (XPA), a nucleotide excision DNA repair disorder with severe neurodegeneration, in silico and in vivo. XPA-deficient cells show defective mitophagy with excessive cleavage of PINK1 and increased mitochondrial membrane potential. The mitochondrial abnormalities appear to be caused by decreased activation of the NAD(+)-SIRT1-PGC-1α axis triggered by hyperactivation of the DNA damage sensor PARP-1. This phenotype is rescued by PARP-1 inhibition or by supplementation with NAD(+) precursors that also rescue the lifespan defect in xpa-1 nematodes. Importantly, this pathogenesis appears common to ataxia-telangiectasia and Cockayne syndrome, two other DNA repair disorders with neurodegeneration, but absent in XPC, a DNA repair disorder without neurodegeneration. Our findings reveal a nuclear-mitochondrial crosstalk that is critical for the maintenance of mitochondrial health.
Overexpression of DNA ligase III in mitochondria protects cells against oxidative stress and improves mitochondrial DNA base excision repair.
Mansour Akbari, Guido Keijzers, Scott Maynard, Morten Scheibye-Knudsen, Claus Desler, Ian D Hickson, Vilhelm A Bohr,
DNA repair - Apr 2014
[abstract]
Base excision repair (BER) is the most prominent DNA repair pathway in human mitochondria. BER also results in a temporary generation of AP-sites, single-strand breaks and nucleotide gaps. Thus, incomplete BER can result in the generation of DNA repair intermediates that can disrupt mitochondrial DNA replication and transcription and generate mutations. We carried out BER analysis in highly purified mitochondrial extracts from human cell lines U2OS and HeLa, and mouse brain using a circular DNA substrate containing a lesion at a specific position. We found that DNA ligation is significantly slower than the preceding mitochondrial BER steps. Overexpression of DNA ligase III in mitochondria improved the rate of overall BER, increased cell survival after menadione induced oxidative stress and reduced autophagy following the inhibition of the mitochondrial electron transport chain complex I by rotenone. Our results suggest that the amount of DNA ligase III in mitochondria may be critical for cell survival following prolonged oxidative stress, and demonstrate a functional link between mitochondrial DNA damage and repair, cell survival upon oxidative stress, and removal of dysfunctional mitochondria by autophagy.
Long-term artificial sweetener acesulfame potassium treatment alters neurometabolic functions in C57BL/6J mice.
Wei-na Cong, Rui Wang, Huan Cai, Caitlin M Daimon, Morten Scheibye-Knudsen, Vilhelm A Bohr, Rebecca Turkin, William H Wood, Kevin G Becker, Ruin Moaddel, Stuart Maudsley, Bronwen Martin,
PloS one - 2013
[abstract]
With the prevalence of obesity, artificial, non-nutritive sweeteners have been widely used as dietary supplements that provide sweet taste without excessive caloric load. In order to better understand the overall actions of artificial sweeteners, especially when they are chronically used, we investigated the peripheral and central nervous system effects of protracted exposure to a widely used artificial sweetener, acesulfame K (ACK). We found that extended ACK exposure (40 weeks) in normal C57BL/6J mice demonstrated a moderate and limited influence on metabolic homeostasis, including altering fasting insulin and leptin levels, pancreatic islet size and lipid levels, without affecting insulin sensitivity and bodyweight. Interestingly, impaired cognitive memory functions (evaluated by Morris Water Maze and Novel Objective Preference tests) were found in ACK-treated C57BL/6J mice, while no differences in motor function and anxiety levels were detected. The generation of an ACK-induced neurological phenotype was associated with metabolic dysregulation (glycolysis inhibition and functional ATP depletion) and neurosynaptic abnormalities (dysregulation of TrkB-mediated BDNF and Akt/Erk-mediated cell growth/survival pathway) in hippocampal neurons. Our data suggest that chronic use of ACK could affect cognitive functions, potentially via altering neuro-metabolic functions in male C57BL/6J mice.
Metformin improves healthspan and lifespan in mice.
Alejandro Martin-Montalvo, Evi M Mercken, Sarah J Mitchell, Hector H Palacios, Patricia L Mote, Morten Scheibye-Knudsen, Ana P Gomes, Theresa M Ward, Robin K Minor, Marie-José Blouin, Matthias Schwab, Michael Pollak, Yongqing Zhang, Yinbing Yu, Kevin G Becker, Vilhelm A Bohr, Donald K Ingram, David A Sinclair, Norman S Wolf, Stephen R Spindler, Michel Bernier, Rafael de Cabo,
Nature communications - 2013
[abstract]
Metformin is a drug commonly prescribed to treat patients with type 2 diabetes. Here we show that long-term treatment with metformin (0.1% w/w in diet) starting at middle age extends healthspan and lifespan in male mice, while a higher dose (1% w/w) was toxic. Treatment with metformin mimics some of the benefits of calorie restriction, such as improved physical performance, increased insulin sensitivity, and reduced low-density lipoprotein and cholesterol levels without a decrease in caloric intake. At a molecular level, metformin increases AMP-activated protein kinase activity and increases antioxidant protection, resulting in reductions in both oxidative damage accumulation and chronic inflammation. Our results indicate that these actions may contribute to the beneficial effects of metformin on healthspan and lifespan. These findings are in agreement with current epidemiological data and raise the possibility of metformin-based interventions to promote healthy aging.
A novel diagnostic tool reveals mitochondrial pathology in human diseases and aging.
Morten Scheibye-Knudsen, Karsten Scheibye-Alsing, Chandrika Canugovi, Deborah L Croteau, Vilhelm A Bohr,
Aging - Mar 2013
[abstract]
The inherent complex and pleiotropic phenotype of mitochondrial diseases poses a significant diagnostic challenge for clinicians as well as an analytical barrier for scientists. To overcome these obstacles we compiled a novel database, www.mitodb.com, containing the clinical features of primary mitochondrial diseases. Based on this we developed a number of qualitative and quantitative measures, enabling us to determine whether a disorder can be characterized as mitochondrial. These included a clustering algorithm, a disease network, a mitochondrial barcode and two scoring algorithms. Using these tools we detected mitochondrial involvement in a number of diseases not previously recorded as mitochondrial. As a proof of principle Cockayne syndrome, ataxia with oculomotor apraxia 1 (AOA1), spinocerebellar ataxia with axonal neuropathy 1 (SCAN1) and ataxia-telangiectasia have recently been shown to have mitochondrial dysfunction and those diseases showed strong association with mitochondrial disorders. We next evaluated mitochondrial involvement in aging and detected two distinct categories of accelerated aging disorders, one of them being associated with mitochondrial dysfunction. Normal aging seemed to associate stronger with the mitochondrial diseases than the non-mitochondrial partially supporting a mitochondrial theory of aging.
Mitochondrial deficiency in Cockayne syndrome.
Morten Scheibye-Knudsen, Deborah L Croteau, Vilhelm A Bohr,
Mechanisms of ageing and development -
[abstract]
Cockayne syndrome is a rare inherited disorder characterized by accelerated aging, cachectic dwarfism and many other features. Recent work has implicated mitochondrial dysfunction in the pathogenesis of this disease. This is particularly interesting since mitochondrial deficiencies are believed to be important in the aging process. In this review, we discuss recent findings of mitochondrial pathology in Cockayne syndrome and suggest possible mechanisms for the mitochondrial dysfunction.
The biarylpyrazole compound AM251 alters mitochondrial physiology via proteolytic degradation of ERRα.
Susan M Krzysik-Walker, Isabel González-Mariscal, Morten Scheibye-Knudsen, Fred E Indig, Michel Bernier,
Molecular pharmacology - Jan 2013
[abstract]
The orphan nuclear receptor estrogen-related receptor alpha (ERRα) directs the transcription of nuclear genes involved in energy homeostasis control and the regulation of mitochondrial mass and function. A crucial role for controlling ERRα-mediated target gene expression has been ascribed to the biarylpyrazole compound 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide (AM251) through direct binding to and destabilization of ERRα protein. Here, we provide evidence that structurally related AM251 analogs also have negative impacts on ERRα protein levels in a cell-type-dependent manner while having no deleterious actions on ERRγ. We show that these off-target cellular effects of AM251 are mediated by proteasomal degradation of nuclear ERRα. Cell treatment with the nuclear export inhibitor leptomycin B did not prevent AM251-induced destabilization of ERRα protein, whereas proteasome inhibition with MG132 stabilized and maintained its DNA-binding function, indicative of ERRα being a target of nuclear proteasomal complexes. NativePAGE analysis revealed that ERRα formed a ∼220-kDa multiprotein nuclear complex that was devoid of ERRγ and the coregulator peroxisome proliferator-activated receptor γ coactivator-1. AM251 induced SUMO-2,3 incorporation in ERRα in conjunction with increased protein kinase C activity, whose activation by phorbol ester also promoted ERRα protein loss. Down-regulation of ERRα by AM251 or small interfering RNA led to increased mitochondria biogenesis while negatively impacting mitochondrial membrane potential. These results reveal a novel molecular mechanism by which AM251 and related compounds alter mitochondrial physiology through destabilization of ERRα.
Sporadic Alzheimer disease fibroblasts display an oxidative stress phenotype.
Mahesh Ramamoorthy, Peter Sykora, Morten Scheibye-Knudsen, Christopher Dunn, Cindy Kasmer, Yongqing Zhang, Kevin G Becker, Deborah L Croteau, Vilhelm A Bohr,
Free radical biology & medicine - Sep 2012
[abstract]
Alzheimer disease (AD) is a major health problem in the United States, affecting one in eight Americans over the age of 65. The number of elderly suffering from AD is expected to continue to increase over the next decade, as the average age of the U.S. population increases. The risk factors for and etiology of AD are not well understood; however, recent studies suggest that exposure to oxidative stress may be a contributing factor. Here, microarray gene expression signatures were compared in AD-patient-derived fibroblasts and normal fibroblasts exposed to hydrogen peroxide or menadione (to simulate conditions of oxidative stress). Using the 23K Illumina cDNA microarray to screen expression of >14,000 human genes, we identified a total of 1017 genes that are chronically up- or downregulated in AD fibroblasts, 215 of which were also differentially expressed in normal human fibroblasts within 12h after exposure to hydrogen peroxide or menadione. Pathway analysis of these 215 genes and their associated pathways revealed cellular functions that may be critically dysregulated by oxidative stress and play a critical role in the etiology and/or pathology of AD. We then examined the AD fibroblasts for the presence of oxidative DNA damage and found increased accumulation of 8-oxo-guanine. These results indicate the possible role of oxidative stress in the gene expression profile seen in AD.
Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy.
Morten Scheibye-Knudsen, Mahesh Ramamoorthy, Peter Sykora, Scott Maynard, Ping-Chang Lin, Robin K Minor, David M Wilson, Marcus Cooper, Richard Spencer, Rafael de Cabo, Deborah L Croteau, Vilhelm A Bohr,
The Journal of experimental medicine - Apr 2012
[abstract]
Cockayne syndrome (CS) is a devastating autosomal recessive disease characterized by neurodegeneration, cachexia, and accelerated aging. 80% of the cases are caused by mutations in the CS complementation group B (CSB) gene known to be involved in DNA repair and transcription. Recent evidence indicates that CSB is present in mitochondria, where it associates with mitochondrial DNA (mtDNA). We report an increase in metabolism in the CSB(m/m) mouse model and CSB-deficient cells. Mitochondrial content is increased in CSB-deficient cells, whereas autophagy is down-regulated, presumably as a result of defects in the recruitment of P62 and mitochondrial ubiquitination. CSB-deficient cells show increased free radical production and an accumulation of damaged mitochondria. Accordingly, treatment with the autophagic stimulators lithium chloride or rapamycin reverses the bioenergetic phenotype of CSB-deficient cells. Our data imply that CSB acts as an mtDNA damage sensor, inducing mitochondrial autophagy in response to stress, and that pharmacological modulators of autophagy are potential treatment options for this accelerated aging phenotype.
SRT1720 improves survival and healthspan of obese mice.
Robin K Minor, Joseph A Baur, Ana P Gomes, Theresa M Ward, Anna Csiszar, Evi M Mercken, Kotb Abdelmohsen, Yu-Kyong Shin, Carles Canto, Morten Scheibye-Knudsen, Melissa Krawczyk, Pablo M Irusta, Alejandro Martín-Montalvo, Basil P Hubbard, Yongqing Zhang, Elin Lehrmann, Alexa A White, Nathan L Price, William R Swindell, Kevin J Pearson, Kevin G Becker, Vilhelm A Bohr, Myriam Gorospe, Josephine M Egan, Mark I Talan, Johan Auwerx, Christoph H Westphal, James L Ellis, Zoltan Ungvari, George P Vlasuk, Peter J Elliott, David A Sinclair, Rafael de Cabo,
Scientific reports - 2011
[abstract]
Sirt1 is an NAD(+)-dependent deacetylase that extends lifespan in lower organisms and improves metabolism and delays the onset of age-related diseases in mammals. Here we show that SRT1720, a synthetic compound that was identified for its ability to activate Sirt1 in vitro, extends both mean and maximum lifespan of adult mice fed a high-fat diet. This lifespan extension is accompanied by health benefits including reduced liver steatosis, increased insulin sensitivity, enhanced locomotor activity and normalization of gene expression profiles and markers of inflammation and apoptosis, all in the absence of any observable toxicity. Using a conditional SIRT1 knockout mouse and specific gene knockdowns we show SRT1720 affects mitochondrial respiration in a Sirt1- and PGC-1α-dependent manner. These findings indicate that SRT1720 has long-term benefits and demonstrate for the first time the feasibility of designing novel molecules that are safe and effective in promoting longevity and preventing multiple age-related diseases in mammals.
Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein.
Michel Bernier, Rajib K Paul, Alejandro Martin-Montalvo, Morten Scheibye-Knudsen, Shaoming Song, Hua-Jun He, Sean M Armour, Basil P Hubbard, Vilhelm A Bohr, Lili Wang, Yaping Zong, David A Sinclair, Rafael de Cabo,
The Journal of biological chemistry - Jun 2011
[abstract]
In mammals, the transcriptional activity of signal transducer and activator of transcription 3 (STAT3) is regulated by the deacetylase SIRT1. However, whether the newly described nongenomic actions of STAT3 toward mitochondrial oxidative phosphorylation are dependent on SIRT1 is unclear. In this study, Sirt1 gene knock-out murine embryonic fibroblast (MEF) cells were used to delineate the role of SIRT1 in the regulation of STAT3 mitochondrial function. Here, we show that STAT3 mRNA and protein levels and the accumulation of serine-phosphorylated STAT3 in mitochondria were increased significantly in Sirt1-KO cells as compared with wild-type MEFs. Various mitochondrial bioenergetic parameters, such as the oxygen consumption rate in cell cultures, enzyme activities of the electron transport chain complexes in isolated mitochondria, and production of ATP and lactate, indicated that Sirt1-KO cells exhibited higher mitochondrial respiration as compared with wild-type MEFs. Two independent approaches, including ectopic expression of SIRT1 and siRNA-mediated knockdown of STAT3, led to reduction in intracellular ATP levels and increased lactate production in Sirt1-KO cells that were approaching those of wild-type controls. Comparison of profiles of phospho-antibody array data indicated that the deletion of SirT1 was accompanied by constitutive activation of the pro-inflammatory NF-κB pathway, which is key for STAT3 induction and increased cellular respiration in Sirt1-KO cells. Thus, SIRT1 appears to be a functional regulator of NF-κB-dependent STAT3 expression that induces mitochondrial biogenesis. These results have implications for understanding the interplay between STAT3 and SIRT1 in pro-inflammatory conditions.
Mitochondrial base excision repair assays.
Scott Maynard, Nadja C de Souza-Pinto, Morten Scheibye-Knudsen, Vilhelm A Bohr,
Methods (San Diego, Calif.) - Aug 2010
[abstract]
The main source of mitochondrial DNA (mtDNA) damage is reactive oxygen species (ROS) generated during normal cellular metabolism. The main mtDNA lesions generated by ROS are base modifications, such as the ubiquitous 8-oxoguanine (8-oxoG) lesion; however, base loss and strand breaks may also occur. Many human diseases are associated with mtDNA mutations and thus maintaining mtDNA integrity is critical. All of these lesions are repaired primarily by the base excision repair (BER) pathway. It is now known that mammalian mitochondria have BER, which, similarly to nuclear BER, is catalyzed by DNA glycosylases, AP endonuclease, DNA polymerase (POLgamma in mitochondria) and DNA ligase. This article outlines procedures for measuring oxidative damage formation and BER in mitochondria, including isolation of mitochondria from tissues and cells, protocols for measuring BER enzyme activities, gene-specific repair assays, chromatographic techniques as well as current optimizations for detecting 8-oxoG lesions in cells by immunofluorescence. Throughout the assay descriptions we will include methodological considerations that may help optimize the assays in terms of resolution and repeatability.
Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane.
Maria D Aamann, Martin M Sorensen, Christina Hvitby, Brian R Berquist, Meltem Muftuoglu, Jingyan Tian, Nadja C de Souza-Pinto, Morten Scheibye-Knudsen, David M Wilson, Tinna Stevnsner, Vilhelm A Bohr,
FASEB journal : official publication of the Federation of American Societies for Experimental Biology - Jul 2010
[abstract]
Cockayne syndrome (CS) is a human premature aging disorder associated with severe developmental deficiencies and neurodegeneration, and phenotypically it resembles some mitochondrial DNA (mtDNA) diseases. Most patients belong to complementation group B, and the CS group B (CSB) protein plays a role in genomic maintenance and transcriptome regulation. By immunocytochemistry, mitochondrial fractionation, and Western blotting, we demonstrate that CSB localizes to mitochondria in different types of cells, with increased mitochondrial distribution following menadione-induced oxidative stress. Moreover, our results suggest that CSB plays a significant role in mitochondrial base excision repair (BER) regulation. In particular, we find reduced 8-oxo-guanine, uracil, and 5-hydroxy-uracil BER incision activities in CSB-deficient cells compared to wild-type cells. This deficiency correlates with deficient association of the BER activities with the mitochondrial inner membrane, suggesting that CSB may participate in the anchoring of the DNA repair complex. Increased mutation frequency in mtDNA of CSB-deficient cells demonstrates functional significance of the presence of CSB in the mitochondria. The results in total suggest that CSB plays a direct role in mitochondrial BER by helping recruit, stabilize, and/or retain BER proteins in repair complexes associated with the inner mitochondrial membrane, perhaps providing a novel basis for understanding the complex phenotype of this debilitating disorder.
Changed mitochondrial function by pre- and/or postpartum diet alterations in sheep.
Wenche Jørgensen, Christiane Gam, Jesper Løvind Andersen, Peter Schjerling, Morten Scheibye-Knudsen, Ole Hartvig Mortensen, Niels Grunnet, Mette Olaf Nielsen, Bjørn Quistorff,
American journal of physiology. Endocrinology and metabolism - Dec 2009
[abstract]
In a sheep model, we investigated diet effects on skeletal muscle mitochondria to look for fetal programming. During pregnancy, ewes were fed normally (N) or were 50% food restricted (L) during the last trimester, and lambs born to these ewes received a normal (N) or a high-fat diet (H) for the first 6 mo of life. We examined mitochondrial function in permeabilized muscle fibers from the lambs at 6 mo of age (adolescence) and after 24 mo of age (adulthood). The postpartum H diet for the lambs induced an approximately 30% increase (P < 0.05) of mitochondrial VO(2max) and an approximately 50% increase (P < 0.05) of the respiratory coupling ratio (RCR) combined with lower levels of UCP3 and PGC-1alpha mRNA levels (P < 0.05). These effects proved to be reversible by a normal diet from 6 to 24 mo of age. However, at 24 mo, a long-term effect of the maternal gestational diet restriction (fetal programming) became evident as a lower VO(2max) (approximately 40%, P < 0.05), a lower state 4 respiration (approximately 40%, P < 0.05), and lower RCR ( approximately 15%, P < 0.05). Both PGC-1alpha and UCP3 mRNA levels were increased (P < 0.05). Two analyzed muscles were affected differently, and muscle rich in type I fibers was more susceptible to fetal programming. We conclude that fetal programming, seen as a reduced VO(2max) in adulthood, results from gestational undernutrition. Postnatal high-fat diet results in a pronounced RCR and VO(2max) increase in adolescence. However, these effects are reversible by diet correction and are not maintained in adulthood.
Regulation of mitochondrial respiration by inorganic phosphate; comparing permeabilized muscle fibers and isolated mitochondria prepared from type-1 and type-2 rat skeletal muscle.
Morten Scheibye-Knudsen, Bjørn Quistorff,
European journal of applied physiology - Jan 2009
[abstract]
ADP is generally accepted as a key regulator of oxygen consumption both in isolated mitochondria and in permeabilized fibers from skeletal muscle. The present study explored inorganic phosphate in a similar regulatory role. Saponin permeabilized fibers and isolated mitochondria from type-I and type-II muscle from male Wistar rats were prepared. Respiration was measured while the medium P(i) concentration was gradually increased. The apparent K(m) values for P(i) were 607 +/- 17 microM and 405 +/- 15 microM (P < 0.0001) for type-I and type-II fibers, respectively. For isolated mitochondria the values were significantly lower than type-1 permeabilized fibers, 338 +/- 130 microM and 235 +/- 30 microM (P < 0.05), but not different with respect to fiber type. The reason for this difference in K(m) values in the permeabilized muscle is unknown, but a similar pattern has been observed for K(m) of ADP. Our data indicate that phosphate may play a role in regulation of oxygen consumption in vitro and in vivo.
See less
The Scheibye-Knudsen lab is currently experiencing significant growth with a reported 2017 increase of 100% percent in lab members. But we are still looking for new members! If you are interested please send an email to mscheibye@sund.ku.dk. We hope to keep this growth up in the coming years.
The crew

Morten Schebye-Knudsen, MD, DMSc.
PI
mscheibye@sund.ku.dk
CV

Guido Keijzers, Ph.D.
Assistant professor
guido@sund.ku.dk

Daniela Bakula, Ph.D.
Post doc
bakula@sund.ku.dk

Garik Mkrtchyan, Ph.D.
Post doc
garik@sund.ku.dk

Brenna Osborne, Ph.D.
Post doc
brenna@sund.ku.dk

Michael Petr
PhD-student
mpetr@sund.ku.dk

Michael Ben-Ezra
PhD-student
mbenezra@sund.ku.dk

Amanuel Teklu
Research assistant
amanuel@sund.ku.dk

Aksel Olesen
Master student
aolesen@sund.ku.dk

Mikkel Dupont
Master student
mdupont@sund.ku.dk

Silja Samuelsen
Master student
silja@sund.ku.dk

Nils Gedsig Kirkelund Madsen
Research assistant
ngkm@sund.ku.dk

Matthew Erickson
Master student
matt@sund.ku.dk

Tulika Sharma
Master student
tulika@sund.ku.dk

Zarah Gertrud Meisen
Master student
zmeisen@sund.ku.dk

Maria Thaysen
Visiting student
mthaysen@sund.ku.dk

Mads Fugl Petersen
Visiting student
madsfp@sund.ku.dk

Mathias Løvgreen Engel
Visiting student
mengel@sund.ku.dk
Karsten Schebye-Alsing, Ph.D.
Benevolent Ingenuity Overlord of informatics
karsten@scheibye-knudsen.dk

A blast from the past when Morten was at the National Institute on Aging, NIH
Aging represents the largest risk factor for most chronic diseases. Notably, the proportion of elderly is increasing across the globe and thus most societies face a significant socio-economic burden of an aging population. Although the cause of aging is unknown recent work suggests that signaling from damaged DNA may be an important factor driving organismal aging.
Importantly, we and others have found that interventions in DNA damage signaling may stall the aging process leading to increased health and longevity. The overall goal of the lab is to further understand the aging process and to develop interventions that may lead to healthier aging.
The goal is pursued through a variety of ways including in silico analyses, in vitro biochemistry and molecular biology as well as in vivo work on mouse models.

DNA damage causes activation of the DNA damage sensor poly-ADP-ribose polymerase 1 (PARP1). PARP1 metabolizes NAD+ and activation of this enzyme thus decrease cellular NAD+ levels. A consequence of loss of NAD+ is decreased production of acetyl-CoA and increased production of lactate.
The consequence of DNA damage is thus less efficient mitochondrial energy production and changes in the global acetylome. Notably, treatment with precursors for NAD+ such as nicotinamide riboside (NR) or ketones leading to increased acetyl-CoA levels. We are interested in understanding the relative role of these changes in the progression of normal and accelerated brain aging.

Work from us and others have demonstrated that targetted interventions at multiple steps in the DNA damage cascade can attenuate features of aging.
This includes inhibition of the DNA damage sensor poly-ADP-ribose polymerase 1 (PARP1), augmentaion of acetyl-CoA levels through ketolysis, boosting NAD+-levels through treatment with nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN). Additionally, boosting DNA repair could be a possible way to attenuate age associated DNA damage although no pharmacological means currently exists to do this. A focus of the lab is to understand and apply these interventions for an aging population.

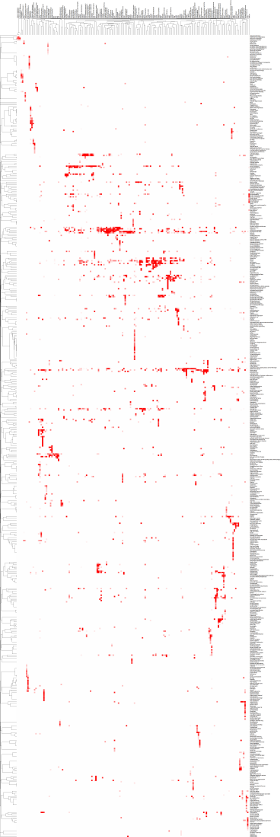
Mitochondrial diseases represent a complex collection of phenotypically diverse disorders with highly variable age-of-onset, clinical presentation and progression. Notably, mitochondrial dysfunction has been proposed to underlie neurodegenerative, cardiovascular and metabolic diseases as well as aging it self. Recently, we have developed a database (www.mitodb.com) that utilizes custom scripts and machine learning algorithms to investigate possible mitochondrial involvement in various diseases. A focus of the lab is to further develop this in silico approach to discover mitochondrial changes that occur in aging and age-associated diseases.
 |
|
The image on the left is a clustermap of how the diseases in the www.mitodb.com database correlates with each other based solely on their traits. Please be patient and wait for the image to finish loading. Each dot represents a referenced trait with the intensity of the color reflecting the prevalence of the trait in that particular disease. Move the cursor over the image to get a closer look. The cluster was made using cluster 3.0. Uncentered similarity metrics and average linkage was used to generate the clustering. |
Several positions are open
The Scheibye-Knudsen lab is looking for highly motivated people interested in aging. Please send your CV to:
contact@scheibye-knudsen.dk

